RasMol is an older program, used for visualizing molecular structures. New users should look to new programs that have built on the idea, but are more convenient and powerful. This page remains for those who already use RasMol, but may need some reminders.
Chime was a browser plug-in to provide RasMol functionality online. It never worked very well, and has been superseded.
|
Basics
A. Introduction B. Getting and installing RasMol C. Getting started D. More small molecules E. Proteins 1. Getting a structure file for a protein 2. Insulin 3. Pokeweed antiviral protein 4. Protein of the month F. Nucleic acids Command Line G. The Command Line H. Measuring distances or angles I. Finding disulfide bonds |
Other
J. Using RasMol (or Chime) online K. Berkeley RasMol Getting started The "Molecules" window Opening multiple files Measuring distances or angles Altering the conformation L. More help M. Making your own 3D computer models Method 1: ACD ChemSketch Method 2: ISIS/Draw and ViewerLite N. Notes for Org/Biochem (X402) class O. Web sites - summary P. Acknowledgments and feedback |
|
| |
RasMol is a program that allows you to view molecular structures on the computer screen, and to manipulate them. RasMol was written by Roger Sayle, now at Glaxo, and is available free, for Windows and Mac computers (and others). (RasMol for Windows is also known as RasWin.)
RasMol was designed for viewing protein structures -- molecules so large that one would not make an ordinary molecular model by hand. However, it can also be used for small molecules. Using RasMol for small molecules is particularly useful if you do not have a set of models. If you do have models, it may be good to learn to use RasMol with small molecules, and even compare the RasMol model with the "physical" models.
This page will help you get started with RasMol. It will guide you through getting and installing RasMol, and getting structure files for viewing in RasMol. It will show you some basic operations, both with small molecules and proteins. (It is not intended to be complete, but only to get you started. I hope it will give you enough that you can continue on your own, with the help of other resources, such as those in the "More help" section below.)
The information here will deal most specifically with RasMol run as a standalone program on a Windows-based computer. However, all the basics are about the same in other cases.
There are also differences between versions of RasMol, which we will largely ignore. Special capabilities of Berkeley RasMol will be discussed later.
RasMol can also be installed as a "helper" in your web browser. Chime is a program with similar functions, which runs as a plug-in in your browser. See the section Using RasMol (or Chime) online, below, for more about these uses.
Structure files for RasMol usually have the file extension .pdb (which stands for protein data bank). RasMol can read some other file formats.
Students in the org/biochem course are encouraged to get RasMol and learn its basic features with small molecules early in the course (Ouellette 2/e Ch 3-4). Later sections of this page deal with proteins; leave these parts until Ch 15. (Chapter references on this page are to the current X402 book, Ouellette 2/e. There is no "need" to have a book in order to use this page. The book references are simply for the convenience of students is a particular course.)
You may find it useful to print out this page.
Go to the RasMol Home Page:
http://www.umass.edu/microbio/rasmol/
To get the RasMol program from this site...
Choose RasMol at the very top of the page.
Scroll down and choose "RasMol Quick Start".
Scroll down and choose "Where do I get RasMol?"
Obtain the version that is appropriate for your system, and install it.
Some people have told me that they are not allowed to "install" programs. For example, they might be using a computer at work, or someone else's computer. For RasMol, this is actually not a serious problem. RasMol does not need to be formally installed. It is ok to just download it to a floppy disk, and run the program from the floppy. The program itself, and nearly all structure files, fit easily on a standard floppy.
If you have access to a computer, but are not able to download files from the Internet... bring me a couple of floppies and I will copy the files for you.
Download the following file for some initial practice with RasMol.
Butane. Simple, straight-chain, saturated hydrocarbon (alkane). Ch 11.
| The butane structure file, above, is from Dr. Dave Woodcock's collection at University of British Columbia (Okanagan). The main address for his collection is https://www.molecularmodels.ca/molecule/molecule_index.html. This site is also listed on the Writing, drawing and viewing chemical formulas page. |
When you try to download a file, your browser may try to open it rather than download it. The details depend on your system. For example, your browser may try to open the file directly in RasMol, within your browser, or may try to open it in Chime. If this is an issue, use a right-button click to directly save the file to your disk. Then choose "Save link as..." (Netscape) or "Save target as..." (Internet Explorer). [In Netscape, SHIFT-click (hold the SHIFT key down while clicking) should also allow you to directly save the file.]
If you want to view pdb files online, see the section Using RasMol (or Chime) online, below.
Now, run RasMol. How you run RasMol depends on your computer system.
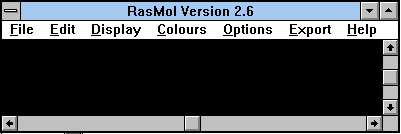
| The following screen shot shows a blank RasMol screen. This is about what you should see when you first load RasMol. (Details may vary. I have greatly reduced the size of the working region here.) |
|
|
Load the butane structure. To load a molecule, go to the File menu, choose Open, then find the file. (Alternatively, you can "drag" the pdb file from any file listing into the open RasMol window.)
At this point, you should see on the RasMol screen something that looks similar to a structural formula for the chemical.
Go to the Display menu, and choose Ball & Stick. The molecule on the screen should now change to something that looks like the ball and stick models we use in class. The C atoms are gray and the H atoms are white. (These colors were also used in the original structure, but you may not have noticed.)
The display you saw originally is called Wireframe. Go to Display menu, and choose Wireframe; the molecule should go back to the form you saw originally. You can switch between Wireframe and Ball & Stick displays at any time, as you wish. You can also experiment with other Display choices. However, some are intended for macromolecules, such as proteins, and will show nothing with a small molecule such as butane.
Here are some basic operations you want to learn:
To rotate the model, around the X and Y axes (the axes in the plane of the screen): Hold the left mouse button down, move mouse (right/left or up/down). This allows you to view the molecule from various orientations. (Also see next command.) For example, it is possible that when you first see the butane on the screen, you can't see all the atoms. By rotating the model, you can reveal features that weren't originally visible.
To rotate the model, around the Z-axis (the axis perpendicular to the screen, towards you): Hold SHIFT and the right mouse button down, move mouse (left/right).
To change the size of the model ("Zoom"): Hold SHIFT and left mouse button, move mouse.
To move the model: Hold right mouse button, move mouse.
Here are a few more files with structures of small molecules, to help you get started. They are listed here, keyed to appropriate chapters of Ouellette 2/e.
In most versions of RasMol, you can only display one molecule at a time. In general, you need to File - Close one structure before opening another. (The "Berkeley" RasMol does allow you to view more than one structure at a time.)
Cyclohexene. Cyclic hydrocarbon, with a double bond (cycloalkene). Ouellette Ch 4.
RasMol may or may not explicitly show double bonds on the screen. It depends on the version of RasMol, and on the particular pdb file. (It may also depend on your particular system.) The current version of RasMol will display double bonds, if the pdb file contains the necessary information. Files at the Okanagan site do now include double bonds. ("Berkeley" RasMol does not show double bonds. In fact, it will execute a command to rotate around a bond that really is a double bond. I think it does not know that the bond is double.)
Benzene. Aromatic ring. Ouellette Ch 5. Rotate this structure so you see the ring "edge on". It is planar.
2-propanol (isopropyl alcohol). Secondary alcohol. Ouellette Ch 8. Oxygen atoms are red.
Thousands of pdb files for small organic and bio-chemicals are available on the web. For some good resources, see the links on the Writing, drawing and viewing chemical formulas page. More sources of pdb files are listed at the RasMol Home Page, described in the section Getting and installing RasMol, above.
| The structure files listed in this section are from Dr. Dave Woodcock's collection at University of British Columbia (Okanagan). The main address for his collection is https://www.molecularmodels.ca/molecule/molecule_index.html. This is one of the sites I refer to in the previous paragraph as a good source of pdb files. |
Structure files (pdb files) for thousands of proteins are maintained at the Protein Data Bank. To get a protein file, go to the pdb home page, at https://www.rcsb.org/.
As an example, get the file for a particular form of insulin -- a small (and well known) protein that we will use in the next part, below. In the search box, type in the code 9ins and then press your Enter key (or click on the search button on the screen).
In this case, I know the code number of a particular file that is useful here. (It is for bovine insulin, and shows the basic insulin "monomer".) In general, you might enter a descriptive term in the search box, such as insulin. If you try that here, look for any mammalian insulin, in monomer form. It would probably be good to avoid mutant proteins at this point. I encourage you to go back and experiment with this search engine.
Choose the desired protein file (9ins, in this case). At the left are options for Display Molecule. Choose RasMol viewer, and download the file.
Load the insulin structure into RasMol, just as you did before.
Reminder. In most versions of RasMol, you can only display one molecule at a time. In general, you need to File - Close one structure before opening another. (The "Berkeley" RasMol does allow you to view more than one structure at a time.)
The appearance should be familiar... a Wireframe structure. The big difference is that the molecule is much bigger (about 500 atoms), so now we will make use of some of the special features of RasMol intended for use with proteins and other macromolecules.
Go to the Colours menu, and choose Chain. You will now see that the two chains of insulin (called A and B chains; Fig 18-3) are distinguished.
[You may notice a tiny third "chain", in red. This is a small cluster of water molecules that was included in the crystal structure. You can see this by switching to Display = Ball & Stick and Colours = CPK. The "red chain" is now seen to have entirely red atoms, which are oxygens. (H atoms are not shown in these structures.) Another way to show that these are waters, using the RasMol Command Line, is described in the section on disulfide bonds, below. If you would like more information about this, or want other ways to explore these waters, check with me.]
Go to the Display menu, and choose Backbone.
Go to the Display menu, and choose Ribbons.
Experiment with the various options suggested above, and also rotate the molecule. These operations will let you begin to see how RasMol helps you visualize features of proteins. Remember that insulin is a very small protein.
I have chosen this protein here because it offers some very nice views of proteins secondary structure. The basic operations suggested in this section are ones you might carry out with any protein.
Obtain a structure file (pdb file) for pokeweed antiviral protein. To do this, follow the instructions above under Getting a structure file for a protein. In this case, use the search term "pokeweed".
Load the file into RasMol.
Go to the Colours menu, and choose Structure.
Red is α-helix; yellow is β-sheet; blue is turns; white is other.
Although the screen display does now identify the types of secondary structure, it is probably hard to see them at this point. There is too much information on the screen. Therefore, let's set the display to a simpler format...
Go to the Display menu, and choose Ribbons.
Now look for α-helical and β-sheet regions. Rotate the structure, to help you see more. (One of the two major secondary structure will become much more apparent upon rotation.)
The Protein Data Bank features one macromolecule (or complex) each month, with pictures and extensive discussion. They call this feature "Molecule of the Month". The archives include... myoglobin, DNA polymerase, nucleosomes, ribosomes, tRNA, activating enzymes.
For the list of featured protein Molecules of the Month: https://pdb101.rcsb.org/motm/
Many structure files are available for a wide range of nucleic acids, sometimes complexed with proteins. A good general source of nucleic acid files for RasMol is the Nucleic Acid Database at http://ndbserver.rutgers.edu/. Follow the links to Nucleic Acid Databases and then Atlas.
In the sections above, we showed that many features of RasMol are available from the menus. Other features are available from the Command Line. This section will show you how to access the Command Line. The following two sections will make use of the Command Line.
It is fine to skip this section until you need it.
Find the RasMol Command Line. Oddly, finding this feature is not always easy, and it does depend on your system. In Windows 95/98, the "RasMol Command Line" should be visible on the Task Bar at the bottom of the screen. In Windows 3.1, go to the Task List, by pressing the Control key and then the Escape key (while still holding the Control key down).
| The following screen shot shows the RasMol Command Line window. In actual use, you will see a blinking cursor just after the Command Line prompt, RasMol> |

|
At the Command Line prompt, you can type commands, followed by the Enter key: Commands are not case-sensitive. Examples will be given in the following sections.
This section will illustrate use of the Command Line to make measurements of molecular features. Specifically, we will measure distances between two atoms, and bond angles.
Load the butane structure. (You can use any structure of your choice here. However, I will provide some specific measurements that are for the butane molecule from the Okanagan site.)
At the Command Line prompt, type the following command, followed by the Enter key:
set picking distance
If you need help finding the Command Line prompt, see the section The Command Line, above. Reminder: Commands are not case-sensitive.
Return to the main RasMol screen. Click on any atom, and then click on any other atom.
Now look at the Command Line window. For each atom you clicked, the Command Line will show some identifying information. Then, after the second atom, it will show the distance between the two atoms, in Angstroms. (1 nanometer = 10 Angstroms.)
If you measure the distance between any two consecutive C atoms in the butane structure, you should get approximately 1.54 Angstroms, the typical C-C bond length in alkanes.
You can measure the distance between consecutive atoms or non-consecutive atoms. Note that the distance between non-consecutive atoms may depend on the particular conformation shown on the screen at the moment.
As a variation, try the command:
set picking monitor
Now, click on any two atoms in succession, just as you did above. Nothing will happen in the Command Line. However, the distance will be marked on the molecule, on a dotted line connecting the two atoms. (This is particularly nice when you measure the distance between two non-consecutive atoms.)
At the Command Line prompt, type the following command:
set picking angle
Return to the main RasMol screen. Click on an atom, then on an adjacent atom, then on another atom adjacent to that one. That is, click on three atoms that define a bond angle.
Now look at the Command Line window. For each atom you clicked, the Command Line will show some identifying information. Then, after the third atom, it will show the bond angle, in degrees.
If you measure any bond angle in the butane structure, you should get approximately 109-110 degrees, the typical tetrahedral bond angle.
The measurement features all do geometric calculations on the structure shown on the screen. Therefore, they are only as accurate as the structures used. For example, many of the pdb files for small molecules may have nominal bond angles, rather than the precise bond angles found in the actual molecules. Further, distances between non-consecutive atoms depend on the conformation.
The measurement features are only in recent versions of RasMol. For Windows, they work in the current version, 2.6beta2, but not with most older versions. For other platforms, let me know if it does not work, and I will provide more notes.
The measurement features are not described in the built-in help files, which are for version 2.5. These features are described in the RasMol Reference Manual. See the More Help section, below, for more about these help files.
The "Berkeley" RasMol will also do these measurements, with a nicer interface.
This section will illustrate use of the Command Line to explore details of protein structure. The specific example here is to show the disulfide bonds in insulin; as part of that, we will mark the cysteine residues.
As a "bonus", we will confirm that the "extra chain" in the insulin display, briefly noted above in the Insulin section, consists of water molecules.
Load the insulin structure.
Then Display Backbone or Ribbons.
And: Colours Chain.
At the Command Line prompt, type the following command, followed by the Enter key:
select cys
If you need help finding the Command Line prompt, see the section The Command Line, above. Reminder: Commands are not case-sensitive.
Return to the main RasMol screen. Go to Colours menu and choose Shapely. You should now see the cysteines colored differently than the rest of the chains (probably yellow). (There are 6 cysteines in insulin. Two of them are adjacent. Thus you should see five "yellow" regions, one of which is twice as big as the others.)
The "select cys" command that you entered above tells subsequent commands or menu choices to act only on that part of the molecule. If you want to return to working on the entire molecule, type the command select all.
Return to the Command Line, and enter the following two commands:
set ssbonds backbone
ssbonds
Return to the main RasMol screen, and you should see three disulfide linkages, each connecting a pair of cysteines. You will need to Rotate the model to see them all clearly. Note that one of the disulfide bonds is intra-chain.
In the Insulin section, above, we noted that the insulin display contained an "extra chain". We also noted that this extra chain consists of water molecules. We can demonstrate that more directly by using the Command Line. Go to the Command Line, and enter the following two commands:
select hoh
color yellowReturn to the main RasMol screen, and you should see that all of the extra features surrounding the protein are now yellow.
The preceding sections use RasMol to view files that are stored on your computer. You can also view pdb files while online, using your Internet browser program. To do this, you need to either:
- install RasMol as a helper program within your Internet browser program
or
- install Chime as a plug-in to your Internet browser program.
How to set these up is beyond the scope of this page, but mainly involves features of your browser. You can obtain Chime from the RasMol home page, which was discussed above in the section Getting and installing RasMol.
RasMol operates the same way within your browser as it does as a standalone program, discussed in the earlier sections. Chime is similar; if a pdb file loads with Chime while you are online, press the right mouse button to reveal some commands, which should look familiar.
Caution... If you have a slow Internet connection and are viewing large files (such as those for macromolecules), it may be more efficient to load each file to your disk, and view it off-line at your convenience.
Berkeley RasMol is a variation of version 2.5 developed at UC Berkeley. It works like the regular versions, with some idiosyncrasies -- mostly good. (Don't worry too much about the warning that comes with it; I have had minimal trouble with it, and certainly it will not damage your system in any way.) This section assumes that you generally know how to use RasMol, and therefore focuses on some special features of Berkeley RasMol.
You can get the Berkeley version of RasMol from the RasMol home page: http://www.umass.edu/microbio/rasmol/. This is the same source discussed above, in the section Getting and installing RasMol.
You can put different versions of RasMol in the same directory (folder), if you want.
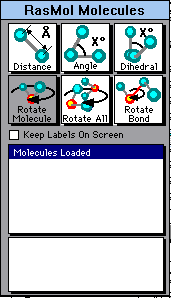
When you run Berkeley RasMol, you will find an extra window -- a small box called the RasMol Molecules window. It will probably first appear right over the main RasMol window. I suggest that you drag it off to the side. (It is fine if it overlaps the main window; you can then easily go between the two windows by clicking on the window you want to be active.)
|
The screen shot at the left shows the RasMol Molecules window. This window is a special feature of Berkeley RasMol. (There are six little boxes in the upper half. Here, five are white and one is shaded. This may vary. More about these boxes later.) You can ignore the Molecules window except when needed for special purposes. That is, Berkeley RasMol runs substantially like other versions, from menus and the Command Line. Additional features are available by using the Molecules window; some of these are discussed in the following subsections. |
With standard versions of RasMol, you can only have one structure file open at a time. (See the section More small molecules, above.) In contrast, Berkeley RasMol allows you to open multiple files at once. This lets you directly compare two molecules on the screen at the same time.
All files that are open are listed in the Molecules Window.
Which molecule is "active", if you give a command? You can choose by clicking on the file name in the Molecules window, or on the structure itself.
You can move the molecules around on the screen, as desired. To do this, make active the molecule you want to move. Then hold down the right mouse button, and drag the structure to wherever you want it.
In the Section Measuring distances or angles, we discussed how to measure distances or angles using the regular versions of RasMol. To do this, you use the RasMol Command Line. Berkeley RasMol will also make these measurements, but using the Molecules window, rather than the Command Line. I think this is easier; at least I would encourage you to try using Berkeley RasMol for these measurements.
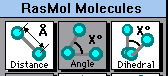
Click on the "Distance" button in the Molecules window. Now click on any two atoms. The distance between them will be shown in the Molecules window, and also on the structure.
| The following screen shot shows the upper part of the RasMol Molecules window. To measure distance between any two atoms (discussed above), click on the left box, labeled Distance. To measure bond angles (discussed below), click on the middle box, labeled Angle. In this picture, the Angle box is shaded, indicating that it has been chosen. |
|
Click on the "Angle" button in the Molecules window. Now click on three consecutive atoms. The angle formed by these three atoms will be shown in the Molecules window, and also on the structure.
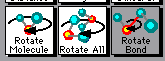
One of the first commands you learn with RasMol is to rotate the molecule, using the left mouse button (Getting started Section, above). Berkeley RasMol offers three variations of the basic rotation command, one of which allows you to alter the conformation of the molecule by rotating part of it around a particular bond. You choose the rotation mode using the boxes in the second row of the Molecules window.
| The following screen shot shows the second row of boxes in the RasMol Molecules window. The three boxes in this row determine how a rotation is done. To rotate part of the molecule around a particular bond, click on the right hand box, labeled Rotate Bond. In this picture, the Rotate Bond box is shaded, indicating that it has been chosen. |
|
The left hand box, Rotate Molecule (which is the default), provides the basic rotate command as before.
The middle box, Rotate All, lets you rotate all the molecules that are on the screen together. (This was not an issue with regular RasMol, since you could only have one molecule open at once.)
The right hand box, Rotate Bond, lets you rotate part of the molecule around a particular bond, thus changing the conformation. To use this, click on the Rotate Bond box, then click on two consecutive atoms, which define a bond. Now carry out the rotate operation, as usual using the left mouse button. You will see the molecule change conformation, around the selected bond. Try it. It's hard to explain, easy to see.
Caution. Berkeley RasMol does not recognize double and triple bonds, even if they are specified in the file. Therefore, if you ask it to rotate around such a bond, it will do so, even though such rotation is not allowed.
RasMol has a regular help file, but you must install it separately. To install the Help files... (In Windows) Get the file rashelp.exe from the RasMol Home Page, which is also listed above under Getting and installing RasMol. Run it, and it will produce two files. These two files (rasmol.hlp and raswin.hlp) must be in the same directory as your RasMol program.
Once you have installed the Help file(s), as discussed above, you can use it from the Help menu (choose User Manual) or from the Command Line prompt (type help or a question mark).
There is also a RasMol Reference Manual in HTML format, which you can save on your computer, and view in your web browser as needed. Apparently, this is the only manual that is completely updated through RasMol version 2.6. It is available from the RasMol Home Page at http://www.umass.edu/microbio/rasmol/distrib/rasman.htm.
More instructional material for RasMol is available at the RasMol Home Page. In particular, you might want to look at two RasMol/Chime FAQ pages there, at http://www.umass.edu/microbio/rasmol/faq.htm.
PDB files simply contain a list of all the atoms, along with their coordinates in space -- their exact position. One way to make a PDB file is to calculate the coordinates. For small molecules, one can use a chemistry "drawing" program to draw the structure, then calculate the coordinates. This section describes two ways to do this, using software that is available for free.
Both methods work fine. If you already have the needed programs, give them a try. If you don't, Method 1 below is probably somewhat simpler. All of the programs are "free" offerings related to commercial software from the companies. All have other features.
See my page ChemSketch - An Introductory Guide for information on getting and using the ChemSketch program, from ACD Labs. The page includes information on viewing the structures in a 3D form, either within a module of that program, or within RasMol.
Method 2: ISIS/Draw and ViewerLite
You will need two programs: ISIS/Draw and ViewerLite.
For information on getting and using ISIS/Draw, see the page ISIS/Draw - An Introductory Guide.
ViewerLite is no longer available from its company. I am not sure it is available now at all. You can look for it if you want; you might search for ViewerLite50.exe. I am leaving this section because it might be useful to some who have the programs. However, if you are starting, it is better to go to ChemSketch, Method 1 immediately above.
The general strategy is to draw the structure in ISIS/Draw, and then have ViewerLite calculate the 3D structure, and save the output as a PDB file. (Isis/Draw has a menu item called "View molecule in RasMol". It seems to not work properly, and we ignore it here.)
The instructions below assume that you have obtained and installed ISIS/Draw and ViewerLite. They also assume basic familiarity with drawing structures in ISIS/Draw. For help with ISIS/Draw, see the page ISIS/Draw - An Introductory Guide. (The required features of ViewerLite are described below.)
To make a PDB file:
1. Draw the desired structure, using ISIS/Draw. Include all the atoms except hydrogens, and show all proper bonding. (ViewerLite will add the H atoms, below.) For help with ISIS/Draw, see the page ISIS/Draw - An Introductory Guide.
Before doing the next step, you need to check a setting in ISIS/Draw. On the Options menu, choose Settings. Then choose the General tab, and turn on the item called Copy Mol/Rxnfile to the Clipboard. (Once you have set it, you shouldn't have to worry about it in the future.)
2. - Select and Copy the structure from ISIS/Draw.
- Paste the structure into ViewerLite.
- Go to step 3, below.
Possible alternative. With some versions of ISIS/Draw and ViewerLite there is an alternative way to get the ISIS drawing into ViewerLite. It is simple, and potentially useful, but seems to not work with some versions. (I have not sorted out the cause, or which is the key program or version.)
To do this...
- Save the structure from ISIS/Draw, as a skc file. This is the default file type from ISIS/Draw, so just Save or Save As, as one would normally do in this program.
- Open the skc file in ViewerLite.
- Go to step 3, below.Check that the resulting 3D model, when viewed in RasMol, has the atoms connected together "reasonably". In some cases, this does not happen. (Errors are obvious, such as atoms bonded to no other atom, or bizarre rings.)
If this method works, it has the advantage of allowing you to use pre-existing SKC files. Try it, but be sure to check that the output is reasonable.
3. Optional -- but probably desired in most cases: put the H atoms in the structure. To do this, go to the Tools menu (of ViewerLite), choose Hydrogens, and then choose Add .
4. Save (Save As) the file (from ViewerLite). Specify the file type as Brookhaven (pdb); check that the file name extension is .pdb.
5. Now try the pdb file you just made in RasMol.
Note that files made this way may have nominal bond angles and lengths, rather than the precise values found in the actual molecules. For example, the bond angle around any C with four atoms attached will be shown as the nominal tetrahedral angle, rather than the actual angles found in that specific molecule. Nevertheless, these nominal structures are useful for seeing the general features -- and it is fun to make your own!
Please let me know of any problems you have with this procedure. Also, please let me know of other ways to make pdb files -- using freely available software.
The general purpose in the sections above was to introduce RasMol. In this section, I will briefly note some specific things that students in the X402 class might look at. Unless noted, all structures for small molecules suggested here can be obtained from Dr. Dave Woodcock's collection at University of British Columbia (Okanagan). The main address for his collection is https://www.molecularmodels.ca/molecule/molecule_index.html. I would like for you to find the files yourself. You can do that by browsing the alphabetical list, browsing the lists by type, or doing a search. I suggest that, over time, you try all these approaches. For proteins, the common source is https://www.rcsb.org/.
In some cases I suggest that you compare two or more structures. There are two ways you might compare two structures on the screen (aside from looking at them one at a time, or printing them out):
As noted above, in the section More small molecules, in most versions of RasMol you can only display one molecule at a time. To view multiple structures together, you can open RasMol twice, and make each a window that fills about half the screen.
The "Berkeley" RasMol does allow you to view more than one structure at a time in the same window.
(Chapter references on this page are to the current X402 book, Ouellette 2/e.)
Most of the comments here refer to resources available at the Okanagan collection.
At the Okanagan site, you can browse lists of compounds by type. There are lists of "alkanes" and (non-aromatic) "cyclic molecules".
We have noted that there is free rotation around single bonds. Nevertheless, at any given instant, the molecule has a particular conformation. (Ouellette 2/e Fig 3.2 etc). Some of these are slightly lower energy than others, because groups are further apart. For large substituents, this can become a significant issue. However, it is largely beyond our course material. The Okanagan collection has three conformations for butane, one of which we used in Section C above to get started. (The conformations of cyclohexane, discussed below, are influenced by the same issues.)
Many derivatives of cycloalkanes are available; one way to find them is to choose the category "Cyclic molecules". The listings include multiple isomers and multiple conformations.
There are many examples of cis vs trans isomers of di-substituted cycloalkanes (Ouellette 2/e Sect 3.5). For example, there are two files for 1,2-diethylcyclobutane, one for each isomer.
There is a special feature on "Cyclohexane conformations", available from the main page. This may be worth exploring. To change the display option... put cursor over a particular structure, right click, and then choose Display. You may want to try Spacefill or Ball and Stick displays.
Check the file listings for cyclohexane. Boat and chair forms are available. The books show the chair form, as generally the most stable conformation (e.g., Ouellette 2/e Fig 3.6 & 7). The boat conformation is an alternative.
For substituted cyclohexanes, conformations are available with the substituent at the axial or at the equatorial position. It might be useful to measure some distances between axial and equatorial groups in cyclohexanes. See the section on Measuring distances or angles if you are using the regular RasMol. The Berkeley RasMol also does this, and maybe more easily.
RasMol may or may not explicitly show double (or triple) bonds on the screen. It depends both on the RasMol version and on the pdb file. Files at the Okanagan site do now include double bonds. The current version of RasMol will display them, but Berkeley RasMol will not. (If you have any trouble with this let me know. If relevant, please give me the pdb file in question.)
An example of an alkene (cyclohexene) was already presented in the More small molecules section, above.
Notice that alkenes are planar around each double bond.
You can find many examples of a pair of cis-trans isomers at the Okanagan collection. A good start would be to look at 2-butene.
Most of the alkenes there are labeled Z or E, rather than cis or trans. The E-Z system for naming alkene isomers is a more complex and more complete system. Ouellette covers it in Sect 4.3. I have placed a brief introduction to it on the page The E-Z system for naming alkenes; examples of using the CIP rules.
Bond lengths and angles. It might be useful -- even fun -- to measure some common bond lengths, and typical bond angles in molecules that are tetrahedral, trigonal planar, and other standard shapes. See the section on Measuring distances or angles if you are using the regular RasMol. The Berkeley RasMol also does this, and maybe more easily.
An example of an aromatic (benzene) was already presented in the More small molecules section, above.
Notice that aromatic rings are planar.
I suggest that you download the structure files for two enantiomers, and view them together. Align the two molecules on the screen so that they are identical for most of the structure. This way you should be able to see one difference -- at the chiral center. A good simple example would be the two enantiomers of 2-butanol. (Note that they are labeled R and S at the Okanagan site. If you haven't mastered the R,S system yet, don't worry about it. It is more important for you to realize they are enantiomers than to know which is which.)
Several sugars are available in the Okanagan collection. For example, you can get structure files for the α and β anomers of D-glucopyranose. Note the name he uses: 'glucopyranose' means glucose in the pyranose (6-membered ring) form. I haven't seen any L-sugars there yet; if you find one, let me know.
You might look at the geometry around atoms with only 2 or 3 atoms attached. For example, when C has 3 bonds (as in carbonyl compounds or alkenes), the geometry is trigonal planar. On the other hand, when N has 3 bonds (amines), the geometry is trigonal pyramid -- because of the "invisible" lone pair on the N.
Structure files for many of the more complex molecules shown in these chapters are available.
In addition to looking at some individual lipid molecules, you might try a tutorial on lipid bilayers, at the RasMol home page. Let me know what you think; I have looked at it only briefly so far. http://www.umass.edu/microbio/rasmol/bilayers.htm
RasMol was designed for proteins. The common file type, pdb = Protein Data Bank, is a clue. The section above on Proteins will get you started. You can then get much more from various resources available at the RasMol home page. The handout listed a web site specializing in amino acids; it is also listed on the Org/BioChem Internet page.
Remember, for finding protein structure files, the common source is https://www.rcsb.org/.
One of the pictures I show in class is of catalase, the enzyme that breaks down hydrogen peroxide. In this tetrameric enzyme (four subunits; quaternary structure), the subunits are actually intertwined. If you want to try to see this, download a file for catalase; be sure to choose one in the tetramer form. Then experiment with display options and rotations to see the intertwining. I suspect that setting Colours to Chain would help. (Caution. File is about 1 Mb.)
See the Writing, drawing and viewing chemical formulas page.
This section lists all external web sites that are mentioned elsewhere on this page, for your convenience.
Links to external sites will open in a new browser window.
http://www.umass.edu/microbio/rasmol/. RasMol home page. Source of RasMol program, plus Help files and manuals, the Chime plug-in, and much more information.
Specific pages there that are mentioned here on my page:
http://www.umass.edu/microbio/rasmol/distrib/rasman.htm. Manual.
http://www.umass.edu/microbio/rasmol/faq.htm. FAQ.
http://www.umass.edu/microbio/rasmol/bilayers.htm. Lipid bilayers.
https://www.molecularmodels.ca/molecule/molecule_index.html. Dr Dave Woodcock's collection of pdb files for small molecules, at University of British Columbia (Okanagan). The page also links to other sources of molecular models and images.
https://www.rcsb.org/. Protein Data Bank.
https://pdb101.rcsb.org/motm/. List of featured protein Molecules of the Month at the Protein Data Bank.
http://ndbserver.rutgers.edu/. Nucleic acid structures. Follow the links to Nucleic Acid Databases and then Atlas.
For help with the drawing programs:
* ChemSketch - An Introductory Guide
* ISIS/Draw - An Introductory Guide.
Special thanks to three people who offered such helpful feedback on an early version of this page: Nick, a high school student in Ontario (the Canadian version); Greg, a university student in England; and Joe, a college instructor here in the Bay Area.
Suggestions and comments are welcomed. Please tell me about any errors you find, as well as suggestions for clarifying or expanding what is here. Contact information is at the bottom of this page.
Writing, drawing and viewing chemical formulas
List of pages of Internet resources
Intro Chem (X11) Home page
Organic/Biochem (X402) Home page Chapter 3 handout [pdf file] Org/BioChem Internet Resources
Molecular biology Home page Molecular biology Internet Resources
Musings (newsletter -- current science)
Contact information Site home page
Last update: August 1, 2022